Features of Molecular Docking Server
You can
- start single molecular docking calculations or high-throughput virtual screening with few clicks
- carry out focused docking to a known binding site
- carry out blind docking experiments to determine possible binding site(s)
- calculate inhibition constants, binding geometry, secondary interactions & much more
- prepare publication quality figures and a method section for your reports automatically
- organize proteins and ligands into user-defined libraries
- carry out molecular docking on-line
Steps of ligand docking
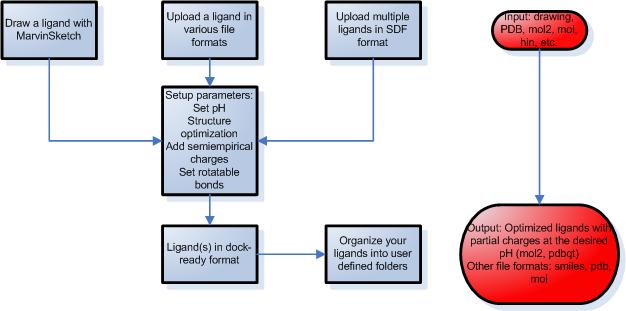
STEP 1 – Preparation of ligands
- Draw your ligands using a Java applet, upload a single ligand file or multiple ligands.
- Draw chemical structures by MarvinSketch, a Java based program with a constantly growing
list of editing features and a number of templates to make molecule drawing simpler.
- Upload a ligand in MDL MOL, SYBYL MOL2, PDB, HYPERCHEM HIN or SMILES format.
- Upload multiple ligands in SDF format.
- You can set various parameters during the simulation such as desired pH, structure optimization
and partial charge calculations using molecular mechanics or semiempirical quantumchemical methods.
- Set up rotatable bonds and atom types automatically or modify manually.
- Download the attached files in several file formats including mol, pdb, mol2 and pdbqt.
- Organize your ligands into self-defined folders. This way the ligands are saved
for later docking calculations.
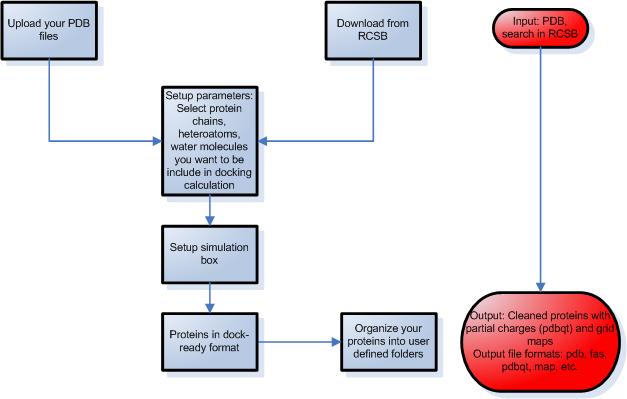
STEP 2 – Preparation of proteins
- Upload protein structures from your files or download them from the
Protein Data Bank using Docking Server by providing the entry code or by text search.
- Select the protein chain, heteroatoms,
ligands and waters present in the protein pdb file that you want to be included
in docking calculation in the process of protein setup.
- Setup the simulation box using one of the following ways:
- select known binding site through a co-crystallized ligand
- select the center of mass of the protein
- select the coordinates of the box center
- select amino acid residues that define the binding site
- Molecular Docking Server calculates necessary map files for each atom type and
prepares the input files for docking calculations.
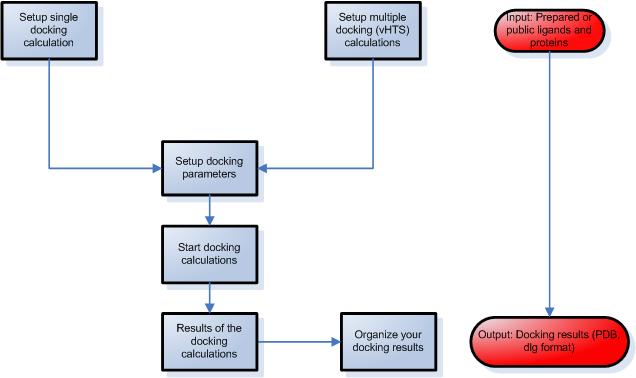
STEP 3 – Setup ligand protein docking calculations
- Select a protein and a ligand from your library.
- Modify advanced parameters during the simulation,
such as number of runs, number of evaluations etc.
STEP 4 – Evalution of results
- Choose an image from the image gallery or render in Molecular Docking Server.
- Analyze the secondary interactions between the protein and ligand.
- Create a method section for your reports automatically.